Chapter 1 Introduction
Phylogenetic biology is the study of evolutionary relationships, and the use of those relationships to study other aspects of biology.
1.1 History of the field
At the dawn of the field, in the second half of the 19th century (Darwin 1859; Haeckel 1897), phylogenetic biology was largely a speculative endeavor. People looked at similarities and differences across organisms, and made hypotheses about the evolutionary relationships that would give rise to those patterns. It wasn’t until the second half of the 20th century that explicit methods were developed to infer phylogenetic relationships (Hennig 1966). This ushered in decades of accelerating phylogenetic methods development that continues to this day. The introduction of explicit model-based approaches to phylogenetic inference was particularly influential (Felsenstein 1981). These new methods were computationally intensive, but fortunately their rise paralleled the rapid development and widespread availability of powerful computers.
Beginning in the late 1980s, molecular data became widely used, based on sequence fragments of a few genes, for building phylogenies of extant organisms. Starting in about 2010, new generations of high-throughput sequencing technology made it possible to collect sequences for thousands of genes from a broad diversity of organisms in a single study (Lemmon and Lemmon 2013), and right now we are in the earliest days of building phylogenies from broadly sampled high-quality chromosome-level genome assemblies.
Phylogenetic biology has always been concerned with figuring out evolutionary relationships. This domain of inquiry is referred to as phylogenetic inference. Phylogenetic inference has been vital to improving the classification of species (phylogenetic taxonomy) (Queiroz 2007) and to reconstructing key features in the evolutionary history of many groups of organisms, and it is of deep interest to those who know the organisms well.
Many questions in biology require knowing evolutionary relationships, even if those evolutionary relationships are themselves not of central interest to the investigator. This has led to the rise of another domain often referred to as phylogenetic comparative biology (Felsenstein 1985b). Instead of ending with trees, as many phylogenetic inference projects do, phylogenetic comparative projects usually start with trees and use them to study the evolution of traits. Comparative questions include whether there is evolutionary covariance between traits, or shifts in rates of evolution along particular branches (also referred to as edges). Phylogenetic comparative methods have become an increasingly large part of the field.
Though phylogeny initially sprang from the field of comparative morphology, and much of the initial focus was on building and analyzing trees with morphological data and using them to study the evolution of morphology, the field has rapidly expanded to encompass many other categories of data. Even so, morphology continues to be vital to building phylogenies in many contexts, including when fossils are available. While most phylogenetic inference is now based on molecular data, phylogenetic comparative methods are routinely applied to all sorts of data and questions. Phylogenetic comparative methods now play important roles in the study of physiology, ecology, genomics, medicine, and most other parts of biology.
Phylogenetics is no longer a strictly macroevolutionary field. Some of the most exciting work in recent years has been at the interface of population genetics and phylogenetic biology, helping to unify our perspectives on micro- and macroevolution. Phylogenies are now also routinely used to study the evolution of genes and other molecular characters, and also to dive into extremely recent and fast evolutionary processes within species, such as viral pandemics.
1.2 Core concepts
There are a few concepts that are fundamental to understanding phylogenetic analyses (Figure 1.1A). These include:
The topology of the phylogeny. This is the structure of the evolutionary relationships. It can be thought of as the branching order of a tree.
The lengths of the branches in the phylogeny. Length can signify different things, such as time elapsed or amount of expected evolutionary change.
Characters. The organism attributes under consideration. These include things like leg length, geographic location, a particular site in a particular gene, a physiological attribute, or a feature of genome organization. Characters are also often called traits.
Character states. The particular values that can be taken by different individuals for specific characters. If leg length is a character, a leg length character state could be 4.3 mm. For a site in a DNA sequence, a character state could be
C,G,T, orA.Models. These are hypotheses about how characters evolve. They take a mathematical and statistical form. For example, a model could describe the covariance between evolutionary changes in different characters, or be a set of functions that describe the probabilities of specific state changes along a branch. Models are abstract representations of biological processes. They are deliberate simplifications that allow us to explicitly describe what we think the most important features of change are. We can make models as simple or complex as we like, but the more complex they are the more information we need to use them effectively.
Model parameter values. These are the specific values for terms in the model. In a model that indicates covariance between characters, the specific values in the covariance matrix are model parameter values. If you have a set of functions that describe the probability of changing between particular states for a character, the model parameters include the rates in those functions.
In order to make informative comparisons across species, both for inferring phylogenies and for phylogenetic comparative analyses, we need a unified ontological framework to refer to the same characters and character states in different organisms. This correspondence is provided by the concept of homology. Homology is a hypothesis that the same attribute is present in different entities because that attribute was present in their shared ancestor. Human arms and bird wings are homologous, for example. Homology is a deceptively simple concept that can sometimes be devilishly difficult to define, test, and apply (Wagner 2014).
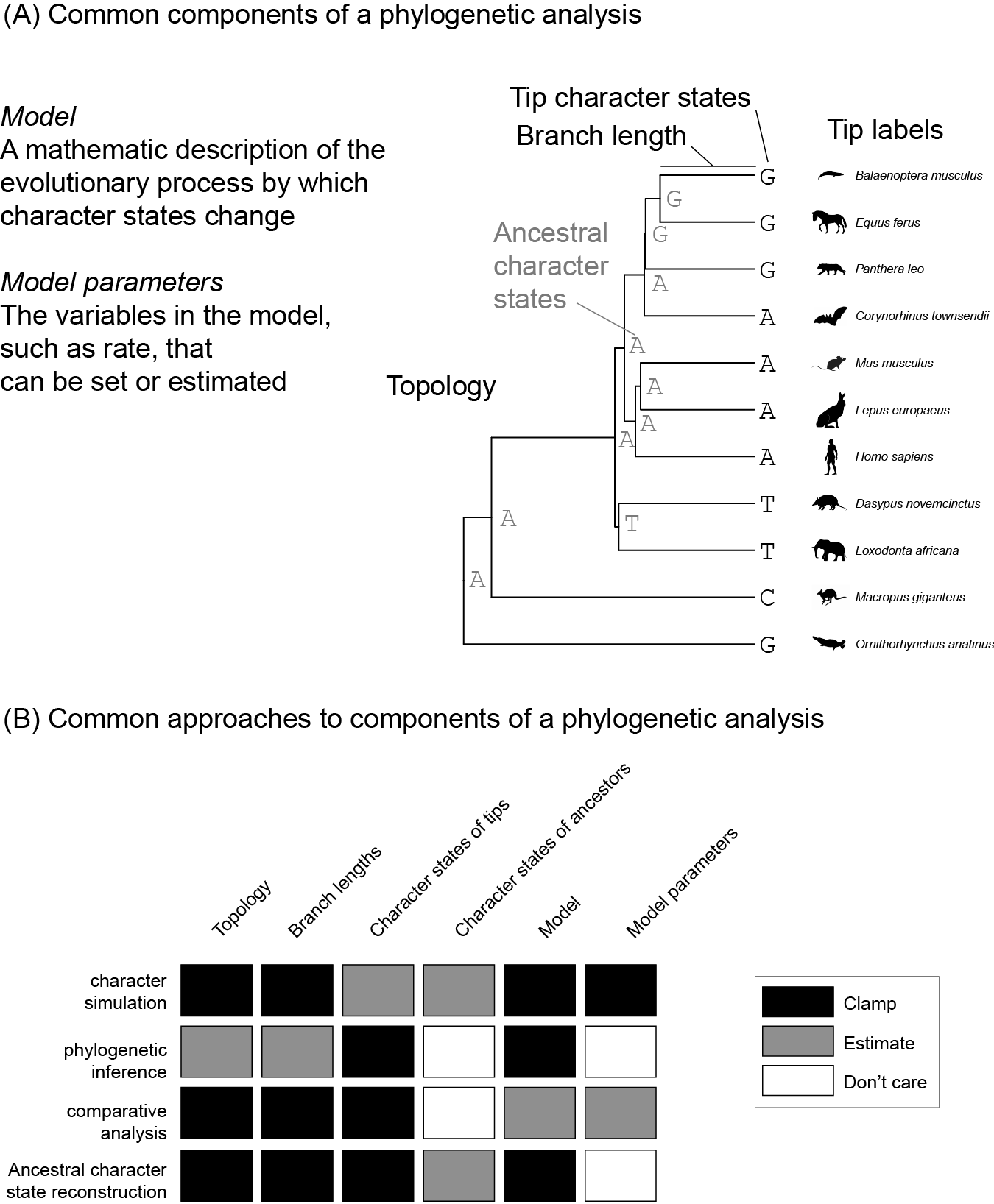
Figure 1.1: (A) The primary components of a phylogenetic analysis. This mammal phylogeny and branch lengths are from http://vertlife.org. The organism silhouettes are from http://phylopic.org/. (B) Different analyses tend to take different approaches to these components.
1.3 A unified perspective on phylogenetic studies
Phylogenetics is such a diverse and quickly growing field that sometimes studies can seem to be more different from each other than they are. This is a shame, as it is a lost opportunity to share ideas across domains in the field and to apply what is learned in one context to another context.
Fortunately, phylogenetic biology has a strong conceptual foundation that provides a unified perspective on phylogenetic studies (Höhna et al. 2014). Once you have this in mind, you can better see the relationships between what may initially appear to be very different methods, questions, and analyses.
Consider some of the concepts we introduced above:
- Topology
- Branch lengths
- Characters
- Character states of tips
- Character states of ancestors
- Model
- Model parameters
The investigator can generally take one of three approaches to each of these (Figure 1.1B). They either:
- Clamp the value according to information in hand, such as their own data or the results of a previous study
- Estimate the value
- Don’t care about the value
Many phylogenetic inference studies use expert knowledge to define and clamp the characters, run some preliminary analyses to evaluate and then clamp the model, and clamp the character states at the tips according to the observed data such as gene sequences. They then estimate the topology, branch lengths, character states of ancestors, and model parameters. They then throw away the estimates of ancestral character states and model parameters, and present the topology and branch lengths as the result. In the end you get a tree of the inferred evolutionary relationships between your organisms of interest.
Many phylogenetic comparative analyses take the same approach to characters and observed character states at the tip (both clamped), and ancestral character states (estimated, but discarded). They then take a different approach to the remaining items. They clamp the tree and branch lengths according to the results of an earlier phylogenetic inference, evaluate different models, and estimate model parameters. The models and model parameters are then the presented results. They would report, for example, that a model that allows for shifts in rates of evolution along different branches is a better fit than a model that doesn’t, or provide estimates of the evolutionary covariance between traits.
Other studies apply the three approaches (clamp, estimate, don’t care) in different ways across the phylogenetic study. Some combinations are very common, others very rare. Some have yet to be explored at all. There are a few reasons for this variation. One is historical. Some combinations became widespread early on and stayed that way as later studies were created in the image of earlier studies. Another reason is methodological. Some combinations are rare or nonexistent in practice because they require methods development that hasn’t been done yet. Data limitations are another big issue. It takes more data to estimate more things. As you get more data, you can unclamp more things. And the biggest limitation is often computational. Phylogenetic analyses are often computationally expensive, and this can quickly become limiting. This is often the ceiling in practice, but more efficient software tools, improved methods, and new data are all helping to make analyses more tractable at the same time that more computer power is becoming available to more investigators.
Once you dig in, the “don’t care” case is surprisingly interesting. Sometimes if you don’t care about something, you don’t need to estimate it. But often you do need to estimate something intermediate to get to what you do care about, and then you just throw away the intermediate result. Things that you aren’t specifically interested in but that you need to estimate to figure out what you do want to learn are referred to as nuisance parameters. Some nuisance parameters aren’t even retained by software, but others are. I mean “don’t care” in the specific sense that these entities aren’t part of your primary question. You should very much care whether your nuisance parameter estimates are sensible since other results depend on them, and you should examine them to make sure they are sensible.
Some studies differ only in which estimates are kept and which are thrown away. One person’s nuisance is another’s bread and butter. One study may keep ancestral character states and discard model parameters, and another study may run essentially the same analysis but discard ancestral character states and keep model parameters.
1.4 Applications
With this framework in mind, let’s take a look at a sampling of recent phylogenetic studies.
Nextstrain provides a frequently updated phylogeny of SARS-CoV-2, with associated data on geography, sampling time, and other factors (Hadfield et al. 2018). This supported genomic epidemiology, surveillance, and public-health decisions during the SARS-CoV-2 pandemic. They are using observed character data at the tips (virus genome sequences) to estimate topology, branch lengths, and model parameters.
Myxozoa are enigmatic parasites that infect fish and invertebrate hosts. They have very few morphological traits that show clear homology with other animals. So Sally Chang, Paulyn Cartwright, and colleagues used phylogenetic inference to examine their relationships to other animals, and found strong support for their placement within Cnidaria (Chang et al. 2015).
My own lab wanted to see if the evolution of gene expression is more rapid after gene duplication than after speciation (Dunn et al. 2018). We found no evidence for such a difference. We used the results of previous studies to clamp gene phylogenies and gene expression character states at the tips, and estimated ancestral expression states to examine shifts in expression along particular branches.
Viburnum is a group of plants with wide distribution in many different habitat types. Michael Landis spearheaded a collaborative project between the Edwards and Donoghue labs here at Yale EEB to understand how Viburnum first diversified in Asia during the Eocene and then spread across the globe (Landis et al. 2021). They clamped the character states at the tips based on gene sequence data, morphology, and geographic range, and developed innovative methods to simultaneously estimate the phylogeny, branch lengths, and ancestral geographic ranges. This is the vanguard of new approaches that simultaneously consider multiple categories of data rather than bolt together multiple independent methods that estimate one thing at a time.
This is a very exciting time in phylogenetic biology. For many years most studies followed a few basic templates. With the development of new phylogenetic methods, new tools to collect high-throughput character data, and a growing interest in phylogenetic questions, the field is now in its most interesting stage of development and application yet.